







Syndrome de Usher, une urgence thérapeutique

Forme la plus courante de surdicécité, le syndrome de Usher fait partie de ces maladies génétiques rares pour lesquelles la science n’a pas encore de traitement. Le point sur l’aspect visuel de cette pathologie avec le Pr Isabelle Audo, PU-PH (Professeur des Universités-Praticien Hospitalier) à Sorbonne Université.

Pr Isabelle Audo, Médecin chercheuse spécialisée dans les maladies rétiniennes génétiques, professeure en ophtalmologie à Sorbonne Université, directrice adjointe de l’Institut de la Vision et coordinatrice du Centre de référence Maladies Rares à l’Hôpital National des 15-20. ©DR
Le syndrome de Usher est une pathologie génétique récessive, qui associe une perte d’audition de profondeur variable jusqu’à la surdité totale, des perturbations de l’oreille interne et des troubles de la vue de type rétinite pigmentaire. Sa prévalence est de l’ordre de 1 sur 10 000 à 1 sur 20 000, 25 000 personnes en France. Un nombre probablement sous-estimé selon le Pr Isabelle Audo : « Je pense qu'il y a peut-être un sous-diagnostic, et qu’on est plus proche de 1 sur 10 000. En effet, même si maintenant, en France, il existe un dépistage périnatal des troubles de l'audition, on ne vérifie pas forcément s’il y a des associations avec des problèmes de vision ». Le syndrome de Usher représente en tout cas plus de 50 % des causes de double handicap visuel et auditif dans les écoles spécialisées pour enfants à handicap. C’est la cause principale de surdicécité en France.
Le Usher, un tableau clinique contrasté
Le Usher se présente sous trois formes cliniques. Le type 1 se manifeste par une surdité profonde voire totale, qui n'est pas compatible avec le développement du langage. En l’absence de dépistage périnatal systématique et d'implantation cochléaire très tôt dans la vie, ce type aboutit à des sujets qui ne développent pas le langage, et sont donc sourd-muet. De plus, ces patients ont une dysfonction vestibulaire qui se traduit par des troubles de l'équilibre. Chez les petits-enfants, ces troubles vont provoquer un retard dans l’acquisition de la position assise, puis de la marche, et des chutes fréquentes. Enfin, les patients présentant ce type clinique développent une rétinopathie pigmentaire qui va être diagnostiquée de façon variable, mais possiblement assez tôt, dans l’enfance.
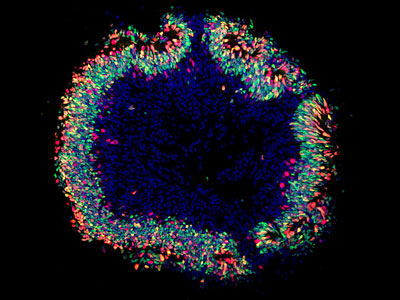
Organoïde rétinien en imagerie de fluorescence. ©Institut de la Vision
Le type 2 traduit une surdité variable, mais en général modérée, compatible avec le développement du langage et stable dans le temps. Il ne présente pas d’atteinte vestibulaire, donc pas de troubles de l’équilibre. La rétinopathie pigmentaire survient en général plus tardivement que dans le type 1. Enfin, dans le type 3, l’ensemble des atteintes est évolutif. C'est-à-dire que ces sujets naissent non sourds ou avec très peu de pertes auditives. Ils vont donc développer le langage. Environ 50 % d’entre eux ont des troubles de l'équilibre, qui vont se développer avec le temps. Ces patients vont devenir progressivement sourds, jusqu’à atteindre parfois la surdité profonde du type 1 à l’âge adulte. Ils développent enfin une rétinite pigmentaire dont la sévérité se situe typiquement entre celles du type 1 et 2.
Syndrome de Usher, le paysage génétique est quasi intégralement décrit
Les trois types cliniques qui ont été décrits sont associés à des causes génétiques différentes. Le travail de l’équipe à l’Institut de l’audition de Christine Petit, professeur à l’Institut Pasteur et au collège de France, a permis d’identifier, sur la base de l’analyse d’une large cohorte européenne, la majeure partie des gènes impliqués dans le syndrome de Usher. « A l’heure actuelle, les gènes que nous connaissons permettent d’expliquer 98% des cas rencontrés. S’il existe encore des causes génétiques à identifier, elles sont extrêmement rares », souligne Isabelle Audo. Ainsi, 4 à 5 gènes différents peuvent être impliqués dans le Usher de type 1, comme dans le type 2. A l’heure actuelle, un seul gène, CLRN1, a pu être relié au type 3. Au sein de ce paysage, certains gènes sont plus fréquemment mis en cause que les autres. Ainsi la majeure partie des cas de type 2 sont dus à des mutations sur le gène USH2A. Quant au type 1, le plus fréquent, dans 70 à 80%, c’est le gène MYO7A qui est en cause. Celui-ci code pour une protéine très, importante pour la structure des photorécepteurs, et qui est également présente dans la couche de soutien de la rétine, l’épithélium pigmenté rétinien. Mais est-ce que la mutation est délétère pour les deux types cellulaires ? Ou impacte-t-elle préférentiellement l’un des deux, sa dégénérescence contribuant au processus pathogénique ? Ces questions, qui restent en suspens, peuvent avoir une conséquence thérapeutique, notamment sur la nécessité de traiter, ou non, les deux types cellulaires.
« Ce qui est assez intriguant, c’est que certaines mutations, selon leur localisation sur le gène en particulier, vont donner soit une surdité isolée, soit une rétinopathie pigmentaire isolée, soit un syndrome de Usher qui se développe secondairement » note la professeure. Cette cartographie, la corrélation phénotype-génotype, le fait qu’une mutation va soit donner un problème juste auditif, soit un problème visuel secondairement, reste encore à définir pour sa plus large part. La comprendre est essentielle car le suivi et la prise en charge des patients ne sont pas du tout les mêmes s’il s’agit d’une forme syndromique, uniquement auditive ou uniquement visuelle. De plus, mieux comprendre le rôle des gènes impliqués, et la façon dont leurs mutations mène au syndrome de Usher peut donner de nouvelles pistes thérapeutiques.

Pr Isabelle Audo ©DR
Syndrome de Usher, la plus grande étude de cohorte française est en cours
Sous l'égide des Pr José-Alain Sahel et Christine Petit, le Pr Isabelle Audo et ses collègues ophtalmologistes, ORL et généticiens ont obtenu la coordination de la partie clinique de Light4Deaf. Lancé il y a presque 10 ans, ce projet de recherche hospitalo-universitaire a mobilisé 13 partenaires publics et privés dont l’Institut de la Vision, la Fondation Voir & Entendre et la Fondation Pour l'Audition. Son objectif était de réunir une grande cohorte de patients atteints du syndrome de Usher, pour les suivre sur 5 ans afin d’améliorer l’état des connaissances au vu des avancées de l'ophtalmologie moderne. « Les techniques d'exploration fonctionnelle ou de structure de la rétine, notamment, sont un petit peu plus précises que ce qu'on avait il y a 30 ans. Cela permet d'avoir une meilleure idée de la progression de cette maladie et de trouver des marqueurs qui évoluent rapidement au cours du temps » détaille le Pr Audo. Ainsi, il est notamment possible actuellement de superposer une analyse de micropérimétrie (un champ visuel très pointu, qui ne fait pas partie de la routine clinique) avec des images de fonds d’œil. Il est donc possible d’identifier des marqueurs qui sont directement corrélés aux anomalies du fond d’œil. Ce sont ces marqueurs qui peuvent, dans le cadre d’un essai clinique, devenir des marqueurs pertinents d'évaluation de l'effet thérapeutique. Si les inclusions dans l’étude sont finies depuis plus de deux ans, les derniers patients sont encore en cours de suivi. En effet, la cohorte réunie pour Light4Deaf regroupe 164 patients, ce qui en fait la plus large en France. A la fin de leur inclusion, de très nombreuses données nécessiteront une analyse minutieuse, avec en ligne de mire une modélisation de la progression de la maladie. « Cet aspect d’évolution, c’est une chose que les patients vont aborder dès les premières consultations. Ils veulent savoir comment la pathologie va progresser, s’ils vont perdre la vue... Les premiers résultats nous montrent que la progression est très difficile à modéliser car il y a une grande variabilité inter-patients », indique le Pr Isabelle Audo. D’où l’intérêt de travailler sur un grand nombre de patients, pour plus de puissance statistique. De plus, souligne la praticienne hospitalière, cette cohorte a permis de constituer un groupe de patients sensibilisés à la recherche clinique, déjà génotypés et prêts à s’impliquer dans des essais de molécules thérapeutiques innovantes.
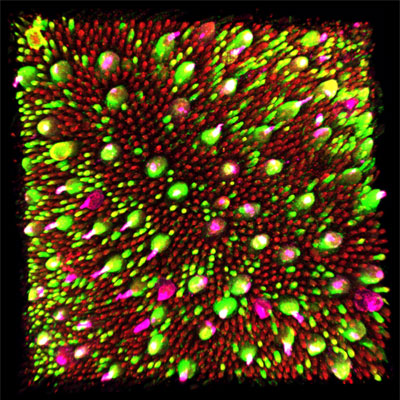
Photorécepteurs. Sabrina El-Yazidi ©DR

Clônes vus en optique adaptative. ©Institut de la Vision
Rendre compte du confort visuel quotidien
Le Pr Isabelle Audo et ses collègues ont obtenu récemment un financement pour continuer cette étude sur un sous-groupe de patients qui ont la forme la plus sévère et aussi la plus fréquente, le syndrome de Usher de type 1 lié au gène MYO7A. Dans ce cadre, les scientifiques cherchent à mettre au point des tests plus à même de rendre compte de l’impact de la pathologie sur la qualité de vie du patient que les simples tests d’acuité et de champ visuel. Pour cela, un partenariat a été mis en place avec la société Streetlab, située au cœur de l’Institut de la Vision, et ses tests de réalité virtuelle, « Nous allons développer des tests de mobilité et les coupler à un questionnaire de qualité de vie, qui nous vient des Etats-Unis, et qui est ciblé sur l’aspect visuel » précise le Pr Isabelle Audo. « L’idée est en effet de tester, en réalité virtuelle, la difficulté de déplacement en condition de pénombre ou de vision nocturne, avec des obstacles plus ou moins contrastés. L’objectif : déterminer, avec les patients, si cette approche permet de mettre en lien leur ressenti, leur quotidien, et les paramètres que les chercheurs veulent évaluer. S’ils se révèlent pertinents, ces tests pourraient permettre de documenter le bénéfice des essais thérapeutiques sur la mobilité ».
A l’heure actuelle en effet, il n’existe pas de traitement pour le syndrome de Usher. Pour les patients atteints du type 1, il existe une prise en charge spécifique, notamment après implantation cochléaire par l’orthophoniste, et une rééducation motrice des troubles de l'équilibre. Dans le standard de soins, les enfants sont pris en charge assez tôt par des psychomotriciens et des kinésithérapeutes qui vont travailler sur les aspects vestibulaire et moteurs. En parallèle, comme pour n'importe quel autre patient atteint de rétinopathie pigmentaire ou de handicap visuel progressif, il va y avoir un travail sur la réhabilitation visuelle. Et lorsque le champ visuel se rétrécit, se mettent en place les cours de locomotion, les cours d'ajustement de la vie quotidienne, l'apprentissage numérique... Mais il n’y a pas de traitement.
Lire la suite de l'article : "Syndrome de Usher : quels espoirs de traitements ?">
Propos recueillis par Aline Aurias
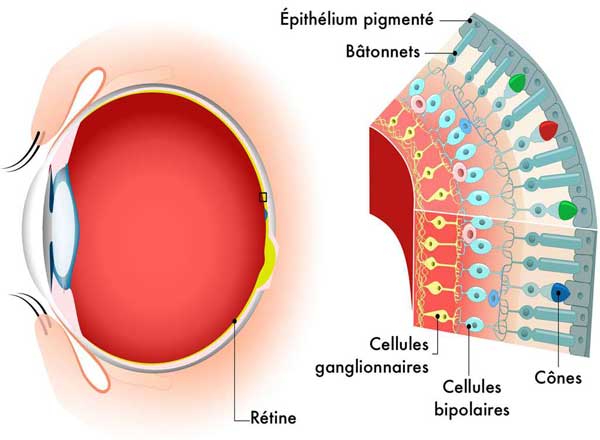
Schéma de l'’œil et coupe de la rétine. ©DR
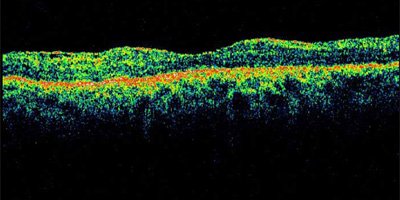
Rétine vue en coupe ©Institut de la Vision
Soutenez la recherche , soutenez l'Institut de la Vision >

À lire aussi

Streetlab, mieux évaluer la vision fonctionnelle pour plus d’autonomie
Créé en 2012, peu après que l’Institut de la Vision soit sorti de terre, Streetlab est une plateforme de recherche qui vient adresser un pan de la vision jusqu’ici resté un peu dans l’ombre : l’évaluation de la perte d’autonomie liée à la déficience visuelle.
Douleur oculaire, une prise en charge unique au CHNO des Quinze-Vingts
La sécheresse oculaire est l’un des premiers motifs de consultation dans les cabinets d’ophtalmologie&nbs

Les troubles de la vision et les maladies des yeux de A à Z

Comment voit l'enfant de la naissance à 6 ans
À sa naissance Dès le 5ème mois de grossesse, la future maman peut sentir le fœtus bouger quand il est soumis à une forte illumination, sur la plage par exemple. Sa naissance... le plus beau jour de votre vie, vous vous demandez si votre bébé vous voit et comment…